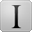脉管性病变(vascular anomolis)是儿童期最常见的病变,总体发病率4%~5%[1]。近年来随着多学科的合作以及基础研究的深入,尤其是1996年国际脉管病变研究协会(International Society for the Study of Vascular Anomalies,ISSVA)推出其分类后,脉管性病变根据血管内皮细胞增生与否分为肿瘤和畸形的观点被广泛接受[2,3]。ISSVA分类的依据主要是临床表现、影像学、血液动力学及病理组织学特点。其最大的优点在于方便临床医师对患者的管理,对治疗有很强的指导意义。2014年ISSVA分类进行修订,分类更细化,同时增加了多种疾病的致病基因及相关的综合征等。
近年来,在多学科尤其是儿科、口腔、整形等学会同行的共同努力下,ISSVA分类在国内逐步被认可和应用,但由于WHO发布的病理分类标准并未将其纳入,因此病理工作者使用ISSVA分类较少,一方面给临床的治疗带来困惑;另一方面也为学科间的交流增加了不便。
脉管性病变大部分发生于婴幼儿,本文对常见于儿童及青少年期的病变进行总结,根据ISSVA分类,分为"肿瘤"和"畸形"两部分,分别描述如下。
又被称为幼年型血管瘤,婴儿富于细胞性血管瘤。是儿童期最常见的肿瘤,占儿童发病率4%~10%[3,4]。在早产低体重儿(体重<1 000 g)中发病率可升高到23%[5]。(1)临床表现:IH好发于女婴,男女比约1∶3~1∶5。肿瘤常见于头部和颈部,其次为躯干和肢体[6]。一般生后2周内出现病灶,也有少部分(约30%)在出生时就有[7]。临床有特殊的生长和消退过程:肿瘤最初为点状红色丘疹或红斑,继而肿瘤迅速生长,称为增殖期,一般在1岁以内;随后肿瘤停止生长,进入为期数年的消退期,表现为颜色变浅,继之出现纵横交错的白色纹理,部分可完全消退[8],大部分病变在7岁以内消退,部分患儿会持续更久一点。最后进入消退完成期,25%~69%的未经治疗的患儿有退行性改变,包括瘢痕、萎缩、色素减退、毛细血管扩张和皮肤松弛[9]。2014年ISSVA分类从3个方面对IH进行了细化,分别是:①根据病变累及部位分为:单发型、多发型、节段型及未定型;②根据病变侵袭程度分为:浅表性、深在性、混合性(浅表+深在)、网状性/顿挫性/微增生性及其他;③合并其他病变:PHACE(后颅凹畸形、血管瘤、动脉病变、心血管病变、眼病变、胸骨裂和/或脐上裂缝)综合征、LUMAR(SACRAL/PELVIS;下半躯体血管瘤、泌尿生殖系统病变、溃疡、脊髓病变、骨畸形、肛门直肠畸形、动脉病变、肾脏病变)综合征。其中节段型IH可能和其他脉管/非脉管畸形的关系尤其密切[10]。(2)病理学改变。增殖期:增生的血管排列成小叶状,小叶间为疏松的纤维组织,小叶内由肥胖的内皮细胞和具有丰富胞质的血管周细胞一起组成毛细血管,管腔小,甚至不明显(图1)。肿瘤性血管可浸润周围组织,如:神经、汗腺等。消退期:小叶内血管腔扩张,内皮细胞及血管周细胞均变扁平,核分裂象明显减少。进而,毛细血管逐渐减少,被纤维脂肪组织取代。间质内可见较多的肥大细胞。消退完成期:病变几乎全部为纤维脂肪组织替代,可伴有瘢痕组织,有时可见少量残留的扩张血管[6]。免疫组织化学:肿瘤性血管内皮细胞特异性表达葡萄糖转运1型(glucose transporter type1,GLUT1)[11],D2-40阴性。
CH是在胎内形成,出生后不再明显进展的肿瘤性病变。根据病变消退的过程,分为:不消退型CH(non-involuting congenital hemangiomas,NICH)[12]、快速消退型CH(rapidaly involuting congenital hemangiomas,RICH)[13]和部分消退型CH(partially involuting congenital hemangiomas,PICH)[14]。目前尚无CH明确的发病率数据,但比IH明显少见[15]。(1)临床表现:CH往往胎儿期超声就被发现,出生即有。无明显性别差异,无种族倾向。最常见的发病部位是头部、颈部和四肢[12,13,16]。和IH相比,RICH的消退出现早、消退速度快,约半数患儿在7个月时肿瘤可完全消退,其余患儿的肿瘤在12~14个月时完全消退。NICH病灶一般不消退,随患儿的生长发育同比例的增大。PICH在出生后开始消退,在消退的过程中可以随时停止,称为部分消退。先天性血管瘤通常表现为单个斑块状或外生性病变,颜色为红色到紫红色,伴有细小或粗大扩张的毛细血管,NICH病变周围皮肤呈环状灰白色晕圈。如伴有溃疡,可能导致严重的出血,甚至危及生命[17]。RICH可引起一过性轻至中度血小板减少,伴/不伴消耗性凝血和高D-二聚体。(2)病理学改变:肿瘤组织位于真皮及真皮下纤维脂肪组织中,由增生的毛细血管排列成大小不等的小叶。小叶内为管径稍大于正常毛细血管的薄壁血管,内皮细胞肥胖,部分细胞核呈鞋钉样凸向管腔。部分小叶中央可见星形扩张的薄壁引流管腔。小叶之间为纤维结缔组织,纤维组织较致密,部分可胶原变性,纤维组织中可见粗大、扩张的营养血管及引流血管(图2)。可见出血、含铁血黄素沉着,局灶见钙化、髓外造血灶等[18]。RICH、NICH或PICH在组织学特征上有重叠,三者区别主要依赖于临床表现。免疫组织化学:肿瘤血管内皮细胞表达CD31、CD34;而GLUT1和D2-40均阴性。
也被译为丛状血管瘤,是一类发生在真皮层内罕见的肿瘤,由变异的毛细血管增生形成具有特征性的簇状/小叶状的结构。目前认为它和卡波西型血管内皮瘤(Kaposiform hemangioendothelioma,KHE)属于同一疾病谱的两个部分,不是独立的病变[10]。(1)临床表现:簇状血管瘤是一种获得性病变,15%为先天性的,大多数发生在5岁以前[19]。无明显的性别差异。肿瘤好发于颈部和躯干的皮肤[20]。皮损表现为孤立的红色或紫色斑块,可伴有皮下结节;也可为弥漫的皮肤色斑[15],部分皮损伴有多毛症。部分病变可自发消退。与KHE相似,簇状血管瘤尤其是先天性簇状血管瘤可引起消耗性凝血病——Kasabach-Merritt综合征(KM综合征)。(2)病理学改变:在真皮层内,增生的毛细血管形成圆形或卵圆形小簇状,以散在、互不连接的"炮弹头"形式分布;小叶内的毛细血管细小,管腔不明显,主要由增生的卵圆形、梭形细胞组成,可见微血栓形成;小叶周边可见扩张的新月形/裂隙样淋巴管样腔隙(图3)。免疫组织化学:肿瘤细胞局灶表达血管内皮标记CD34及淋巴管内皮标记D2-40,阴性表达GLUT1[3]。
是由不成熟血管构成的肿瘤,属于交界性/中间型肿瘤。此瘤罕见,发病率每10万名儿童中不到1人[21]。(1)临床表现:KHE常见于婴幼儿,大部分发病于2岁以前,也有先天性患者或发生于成年人的报道[21]。无明显性别和种族差异,临床可伴有KM综合征[22,23]。多发于后腹膜和皮下以及深部软组织。KHE可以并发淋巴管瘤病[22]或先天性淋巴水肿(Milroy disease),也可在脉管畸形基础上发生。皮肤病变表现为境界不清的紫红色斑块,累及皮肤全层并向皮下组织延伸。皮肤表面的出血症状有时可以作为KM综合征的早期指征。深部软组织病变表现为单个或多个肿块,可累及下方骨组织,少数累及淋巴结[24]。本病可局部侵袭性生长,但无远处转移的报道。与簇状血管瘤相比,KHE更易引起KM综合征,尤其病变直径>8 cm;婴儿期出现;以及累及内脏和/或肌肉的肿瘤[21]。(2)病理学改变:典型病变由数个边界不清、浸润生长的实性结节组成。肿瘤的实性结节由梭形细胞和小血管腔组成,形成新月形或鱼嘴样腔隙(图4)。细胞无明显异型性,可见核分裂象。有时可见肿瘤细胞增生形成"具有肾小球样外观的"细胞簇。部分管腔或细胞内可见含铁血黄素沉着,结节周围的小血管中常常含有微血栓。结节间为纤维结缔组织,少量病例可见纤维化区域和伊红色无结构石棉样纤维区域[25]。部分KHE病例可伴发异常淋巴管或淋巴管瘤病[26],KHE与淋巴管瘤病区域可分界清楚或混杂分布。免疫组织化学:KHE具有增殖期内皮细胞的特征,部分具有淋巴管上皮分化趋势。阳性表达CD31、CD34和D2-40[22,25,27,28],人类疱疹病毒8(HHV8)、平滑肌肌动蛋白(SMA)和GLUT1阴性。
属于血管反应性增生还是肿瘤性增生尚有争议[29]。ISSVA目前将其列入良性肿瘤[10]。(1)临床表现:化脓性肉芽肿可见于儿童和成年人。在儿童期,男孩多见。病变常见于头、颈部,一般累及皮肤(约88%),少部分发生于黏膜(约12%)[15]。肿瘤一般较小,表现为紫红色息肉样的单发病灶,表面可有出血或溃疡,直径大都在1 cm以内。也可发生在其他脉管病变的基础上。(2)病理学改变:典型的病变表现为毛细血管增生,排列成小叶状。常发生于黏膜或真皮内,呈外生性。小叶内为增生的毛细血管、小静脉及其他小血管,内皮细胞肥胖,可见核分裂象。小叶间为纤维结缔组织。间质可水肿伴有炎性细胞浸润。大部分病变上方的黏膜或皮肤有溃疡形成,溃疡周边的鳞状上皮增生,尤其是围绕病变的两侧及底部,呈"衣领状"增生(图5)。免疫组织化学:肿瘤细胞阳性表达CD34和CD31;GLUT1和D2-40阴性。
以前被称为梭形细胞血管内皮瘤,被认为具有低度恶性潜能,随后的研究显示其属于良性的反应性病变[30,31]。ISSVA将其列入良性肿瘤[10]。(1)临床表现:梭形细胞血管瘤是一种生长缓慢的良性血管病变,常见于青少年和年轻人。无性别倾向。病变与Maffucci综合征、Klippel-Trénaunay综合征和其他血管畸形相关。好发于四肢远端[30]。大部分为皮肤或皮下单个结节,少部分可多发,但病灶多分布在同一区域[32]。(2)病理学改变:病变位于真皮或皮下,边界清,无包膜,由明显扩张的薄壁血管和实性的梭形细胞两种区域组成(图6)。部分扩张的血管腔内可见血栓;血管间为实性的增生的短梭形、梭形细胞,间质内常见外渗的红细胞,类似于卡波西肉瘤结节期图像;在实性细胞区域另可见小簇状分布圆形、卵圆形胞质空泡状细胞。免疫组织化学:血管内皮细胞阳性表达CD31、CD34;梭形细胞阳性表达SMA、D2-40和Prox1[33]。
除以上类型外,ISSVA分类中还包括一些少见的,或在儿童期少见的肿瘤,主要包括上皮样血管瘤(epithelioid hemangioma)、网状血管内皮瘤(retiform hemangioendothelioma)、乳头状淋巴管内血管内皮瘤(papillary intralymphatic angioendothelioma,PILA或Dabska瘤)、上皮样血管内皮瘤(epithelioid hemangioendothelioma,EHE)和血管肉瘤(angiosarcoma)。另外,除皮肤及软组织外,一些脏器尤其是肝脏的血管瘤也有其各自特点,本共识不展开赘述。
脉管畸形是胚胎发育过程中因血管淋巴管发育异常,血管淋巴管过度增长形成的病变[34]。脉管畸形出生时即存在,大部分可以被发现,少部分在幼年或青少年时才被发现。其生长速度与身体生长基本同步,一般不会自行消退。与血管瘤最大的区别是没有血管内皮细胞的异常增生。在ISSVA分类中脉管畸形分为4组[10]:单纯性畸形、混合性畸形、主要大血管畸形及与血管病变相关综合征。
主要由一种脉管构成,包括毛细血管畸形、静脉畸形、动静脉畸形、淋巴管畸形。
1.毛细血管畸形(capillary malformation)。(1)临床表现:毛细血管畸形表现为皮肤或黏膜粉红色到红色斑片,压之褪色或不完全褪色。最常见的是葡萄酒色斑(port-wine stains,PWS),又称鲜红斑痣,系先天性皮肤毛细血管扩张畸形,发病率约0.3%[35]。毛细血管畸形出生时就存在,并持续一生。随着年龄的增长,病灶颜色逐渐加深、增厚,并出现结节样改变[3]。部分严重的病变可伴有软组织或骨组织的增生,导致患部增大变形等。一些位于头部的中线(额头、眼皮或眉间,也称为天使之吻或鹳咬)或颈背的毛细血管畸形可以减轻和消失。(2)病理学改变:婴儿期毛细血管畸形表现为真皮浅层毛细血管和毛细血管后微静脉的扩张(图7),病变性质是非增生性的,管壁由单层内皮细胞构成,表皮及周围组织正常。随着年龄增长,血管壁增厚,临床表现为病灶颜色逐渐加深、增厚,并出现结节样增生。免疫组织化学:表达CD31、CD34、Fli-1、第八因子相关抗原(FⅧ)等血管内皮细胞标记,不表达GLUT1。
2.静脉畸形(venous malformation)。(1)临床表现:静脉畸形以往称为海绵状血管瘤,是静脉异常发育产生的血管结构畸形。静脉畸形出生时即存在,大部分可以被发现,少部分在幼年或青少年时才被发现。好发部位为头、颈、颌面,四肢、躯干次之。其生长速度与身体生长基本同步,不会自行退化,发病无性别差异。静脉畸形表现为柔软、可压缩、无搏动的包块。包块大小可随体位改变发生变化。表面皮肤可以正常或深蓝色。由于畸形血管的血流缓慢,可能发生静脉血栓,表现为疼痛和触痛。有时可触及静脉石,影像可见钙化。因血液淤滞也可以造成消耗性凝血病。瘤体逐渐生长增大后,可引起沉重感和隐痛。(2)病理学改变:形态学上,静脉畸形可以是局灶性、多灶性或弥漫性,弥漫性的病变常累及整个肌肉或肢体。镜下表现为大小不等的扩张的血管腔窦,内衬正常扁平的内皮细胞(图8),内皮细胞下为一单层基底膜。管腔壁平滑肌稀少,外膜纤维变性。静脉畸形通常为单一的静脉结构,也可与其他脉管结构混合存在形成混合性脉管畸形。免疫组织化学:表达CD31、CD34、Fli-1、FⅧ等血管内皮细胞标记,不表达GLUT1。
球静脉畸形是特殊的静脉畸形。血管球细胞起源的病变被描述为实性型、弥漫型、孤立型、多发型、成人型、儿童型等多种类型。目前认为这些病变分为两大类:(1)固有球瘤(glomus tumor proper):往往是孤立的、界限清楚的实性肿块;由形态单一的圆形细胞构成,属肿瘤性病变。(2)球静脉畸形(glomuvenous malformation,GVM):占血管球细胞病变的20%,以前这类病变也被称为球血管瘤[3,36](glomangioma或glomangiomatosis),通常是位于真皮及皮下的多灶性病变,在皮肤形成结节样、斑块样或鹅卵石样改变,罕见于黏膜组织。皮肤通常是深蓝色到紫色,可压缩性比普通静脉畸形小,触痛比成人型血管球瘤少。组织学上界限不清,病变内见扩张的薄壁静脉,静脉壁周围见一层或多层立方的"球细胞"衬覆(图9,图10)。这些球细胞常呈结节状分布,所以必须充分取材。现在认为这类病变叫球静脉畸形更符合病变的性质[36,37]。
3.动静脉畸形(arteriovenous malformation)。(1)临床表现:动静脉畸形以往称蔓状血管瘤,源于胚胎第4~8周血管发育缺陷畸形[38]。发病率低,无性别差异。40%~60%的患者出生时即发现,易被误诊为毛细血管畸形或血管瘤。好发部位为头颈部,其次为四肢、躯干和内脏。表现为皮肤红斑、皮温高、可触及搏动或振颤。还可以出现疼痛、溃疡、反复出血,甚至心力衰竭。动静脉畸形还可引起外观畸形、重要组织器官受压、功能损害等。Schobinger将动静脉畸形分为4期:Ⅰ期为静止期,Ⅱ期为扩张期,Ⅲ期为破坏期,Ⅳ期为失代偿期。(2)病理学改变:动静脉畸形是高流量的先天性血管畸形,由扩张的动脉和静脉组成(图11),异常的动静脉之间缺乏正常毛细血管床。免疫组织化学:表达CD31、CD34、Fli-1、FⅧ等血管内皮细胞标志物,不表达GLUT1。
4.淋巴管畸形(lymphatic malformation)。(1)临床表现:淋巴管畸形以往称为"淋巴管瘤",是一种常见的先天性脉管畸形。淋巴管畸形的发病率为1/4 000~1/2 000[39]。淋巴管畸形多在2岁前发病,约50%患者出生时即可发现。发病部位以颈部及腋下最常见,腹股沟、纵隔、腹膜后次之,躯干及四肢也可见。淋巴管畸形由一个或多个大小不等的囊腔组成,囊腔之间可以相通或不通。囊内含透明液体,也可呈琥珀色,有波动感。微囊型淋巴管畸形病灶相对较实性。淋巴管畸形的临床表现受病变的类型、范围和深度的影响差异很大,从皮肤黏膜上充满液体的小泡到巨大的肿物。皮肤颜色一般正常,当病灶内出血时皮肤颜色可为暗红色。(2)病理学改变:淋巴管畸形可能源于脉管系统发育早期异常生长和扩张的畸形组织。镜下由大小不等、形状不规则、壁薄的淋巴管腔构成,内衬淋巴管内皮,腔内充满淋巴液,周围有成纤维细胞、白细胞、脂肪细胞和肌细胞等。但在淋巴管畸形的整个病理过程中,没有淋巴管内皮细胞数量的增加,也没有内皮细胞形态和功能的异常,仅是淋巴管管腔直径变化,结构紊乱。淋巴管畸形分为大囊型、微囊型和混合型3型。大囊型由一个或多个大囊腔构成(以往称为囊肿型或囊性水瘤),而微囊型淋巴管畸形(图12,图13)则由多个体积较小的囊腔构成(以往称为毛细管型和海绵型),二者都有的称为混合型淋巴管畸形。大囊型和微囊型的囊腔直径没有统一标准,目前认为>1 cm为大囊型,<1 cm为微囊型[10,39]。免疫组织化学:表达CD31、D2-40,不表达GLUT1。
混合性脉管畸形由两种或两种以上的脉管构成(除外动静脉畸形)。其临床表现依据畸形的成分而不同。常见的混合性脉管畸形类型,毛细血管-静脉畸形(CM+VM)、毛细血管-淋巴管畸形(CM+LM)、毛细血管-动静脉畸形(CM+AVM)、淋巴管-静脉畸形(LM+VM)、毛细血管-淋巴管-静脉畸形(CM+LM+VM)、毛细血管-淋巴管-动静脉畸形(CM+LM+AVM)、毛细血管-静脉-动静脉畸形(CM+VM+AVM)、毛细血管-淋巴管-静脉-动静脉畸形(CM+LM+VM+AVM)。
病变的脉管包括淋巴管、静脉及动脉,畸形类别包括起源、走行、数量、长度、直径(发育不全、过度发育、膨胀/动脉瘤)、瓣膜的异常以及动静脉瘘和胚胎血管持续存在。
血管畸形常合并其他畸形,常见的血管畸形相关综合征见表1。
血管畸形合并其他病变
血管畸形合并其他病变
| 病变名称 | 特征 |
|---|---|
| Klippel-Trenaunay综合征 | 毛细血管畸形,静脉畸形(伴或不伴有),淋巴管畸形,肢体过度发育 |
| Parkes-Weber综合征 | 毛细血管畸形,动静脉瘘,肢体过度发育 |
| Servelle-Martorell综合征 | 肢体静脉畸形,骨骼生长不良 |
| Sturge-Weber综合征 | 面部及软脑膜的毛细血管畸形,眼部畸形(伴或不伴有),骨和/或软组织过度生长 |
| 四肢毛细血管畸形+先天性非进行性肢体过度发育 | |
| Maffucci综合征 | 静脉畸形(伴或不伴有),梭形细胞血管瘤,内生软骨瘤 |
| 巨头-毛细血管畸形(M-CM/MCAP) | |
| 小头-毛细血管畸形(MICCAP) | |
| CLOVES综合征 | 淋巴管畸形,静脉畸形,毛细血管畸形(伴或不伴有),动静脉畸形,过度生长的脂肪瘤 |
| Proteus综合征 | 毛细血管畸形,静脉畸形和/或淋巴管畸形,不对称性躯体过度发育 |
| Bannayan-Riley-Ruvalcaba综合征 | 动静脉畸形,静脉畸形,巨头畸形,过度生长的脂肪瘤 |
| CLAPO综合征 | 下唇毛细血管畸形,面颈部淋巴管畸形,不对称部分或广泛过度生长 |
血管畸形相关综合征这类病变根据病情做必要的辅助检查,明确病变累及的范围、深度及与周围组织的关系,辅助检查包括B超、MRI、CT、眼科检查、听力检查、脑电图及智力筛查等。治疗原则:对病情进行充分评估,选择合理的治疗方案。
脉管病变包括真性血管瘤和脉管畸形两大类,种类繁多,治疗手段也不同。即使同一类型,由于病变部位、范围等的不同,治疗方案也有区别。如婴儿型血管瘤,浅表型一般用普萘洛尔等β受体阻滞剂治疗有效,而混合型除用β受体阻滞剂外可能还要根据实际情况辅助局部注射或手术治疗方能达到较满意疗效。脉管畸形以头颈部多见,可导致明显的外观畸形和器官移位,若影响邻近的骨骼或关节,可引起骨骼变形肥大,导致疼痛及功能障碍。发生在肢体和躯干静脉畸形会形成血栓,导致疼痛及功能障碍。因此,对于头面部和伴有疼痛的肢体病灶,应尽早治疗。治疗方法可选择激光、注射硬化剂(无水乙醇、博莱霉素、泡沫硬化剂等)及手术治疗[3,9,38,40,41,42]。总之,脉管病变的治疗原则是在规范诊断的基础上,进行个体化治疗。
由于脉管性病变诊治涉及多个学科,统一的标准,才能完善诊疗规范。而目前国际、国内均以ISSVA诊断分类标准作为基础,因此病理医师应提高对ISSVA分类的认识,准确的分类才能指导临床选择合适的治疗方案,避免过度治疗或不当治疗。
参与编写的专家名单(以单位名称汉语拼音字母顺序排列)北京大学医学部(王华),重庆医科大学(徐曼),大连市妇女儿童医疗中心(朴正爱),福建医科大学附属协和医院(康德勇),复旦大学附属儿科医院(陈莲),广东省妇幼保健院(郜红艺),广州市妇女儿童医疗中心(王凤华),哈尔滨医科大学附属第二医院(陈英准),哈尔滨医科大学附属第四医院(张淑君),哈尔滨医科大学附属第一医院(姜杰),河北省儿童医院(安会波),河北医科大学第二医院(吴文新),湖南省儿童医院(陈卫坚),江西省儿童医院(杨文萍),解放军北部战区总医院(明健),陆军军医大学第一附属医院(平轶芳),南京医科大学附属儿童医院(武海燕),山东大学齐鲁儿童医院(刘秀美),山西省儿童医院(赵文英),上海交通大学医学院附属上海儿童医学中心(殷敏智),上海交通大学医学院附属新华医院(吴湘如),首都儿科研究所(邹继珍),首都医科大学附属北京安贞医院(陈东),首都医科大学附属北京儿童医院(何乐健),四川大学华西医院(姜勇),苏州大学附属儿童医院(朱雪明),天津市儿童医院(胡晓丽),温州医科大学附属育英儿童医院(谢小志),武汉市妇女儿童保健中心(张文),西安市儿童医院(陈广生),新疆医科大学第一附属医院(李新霞),浙江大学医学院附属儿童医院(汤宏峰),郑州大学第三附属医院(雷冬梅),郑州大学第一附属医院(马怡晖),中国医科大学附属盛京医院(吕庆杰),中国医学科学院北京协和医学院北京协和医院(卢朝辉),中南大学湘雅医院(肖德胜)
执笔人:殷敏智(上海交通大学医学院附属上海儿童医学中心病理科200127);武海燕(南京医科大学附属儿童医院病理科210008)
利益冲突 所有作者均声明不存在利益冲突